Damon Runyon News
View New Articles By
View New Articles By
The Damon Runyon Cancer Research Foundation is thrilled to continue our partnership with the Timmerman Traverse, an adventurous initiative that brings leaders and investors in biotech together to scale extraordinary heights.
The University of Texas MD Anderson Cancer Center, where former Damon Runyon Clinical Investigator Cassian Yee, MD, runs his lab, is home to the Moon Shots program, a cancer research initiative inspired by America's drive toward space in the 1960s. Recently, Dr. Yee and his colleagues announced a project that combines these two ambitions: sending T cells into space to inform the development of new cancer treatments.
In cancer treatment, “targeted therapies” refer to drugs that identify and attack specific proteins in cancer cells that help them survive and grow, while leaving normal cells alone. Due to their specificity, targeted therapies tend to be less toxic than chemotherapy or radiation therapies. Often, they take the form of small molecule inhibitors, which bind to the cancer-promoting proteins and disable them. Unfortunately, however, small molecule inhibitors only work for a subset of cancers.
Although many childhood cancers are now curable with chemotherapy, these lifesaving treatments often carry serious long-term side effects. Studies have shown, for example, that childhood cancer survivors are fifteen times more likely than the general population to suffer from congestive heart failure. For patients and pediatric oncologists, the toxicity of chemotherapy drugs is tolerated only because there are no better options—in the United States, that is.
Each year, the Damon Runyon-Jake Wetchler Award for Pediatric Innovation is given to a third-year Damon Runyon Fellow whose research has the greatest potential to impact the prevention, diagnosis, or treatment of pediatric cancer. This year, the award recognizes the work of Yapeng Su, PhD, a Damon Runyon Quantitative Biology Fellow at Fred Hutchinson Cancer Research Center in Seattle.
Adoptive T cell therapies, in which a patient’s own immune cells are genetically engineered to target their cancer cells, have been remarkably effective in treating certain blood cancers. Unfortunately, this success has not translated to solid tumors, where T cells face unique challenges in the tumor environment that limit their persistence and function.
Few scientific studies meet with more controversy than those that suggest a substance may cause or prevent cancer. As a leading epidemiologist of colorectal cancer, former Damon Runyon Clinical Investigator Andrew T. Chan, MD, MPH, is no stranger to this rollercoaster.
Lung cancer is the leading cause of cancer death in the United States, and nearly a third of these cancers are driven by mutations in the KRAS gene. Long considered an “undruggable” cancer target, mutant KRAS proteins are known to rewire alveolar type II progenitor (AT2) cells, which line the lung surface and are responsible for repairing lung tissue after injury. KRAS inhibitors are now making their way to the clinic, but as yet only a subset of patients respond, highlighting the need to better understand the role of mutant KRAS in the development of lung cancer.
The Damon Runyon Cancer Research Foundation has named 16 new Damon Runyon Fellows, exceptional postdoctoral scientists conducting basic and translational cancer research in the laboratories of leading senior investigators. This prestigious Fellowship encourages the nation's most promising young scientists to pursue careers in cancer research by providing them with independent funding ($300,000 total) to investigate cancer causes, mechanisms, therapies, and prevention.
Some cancer cells, such as those in lung tumors, change drastically in appearance and behavior when they develop resistance to targeted therapies. The result of these changes, collectively known as histological transformation (HT), is a more aggressive tumor type. HT necessitates a new therapeutic strategy, since the original oncogene is no longer driving the tumor’s spread. But first, researchers have to find out which genes have assumed control.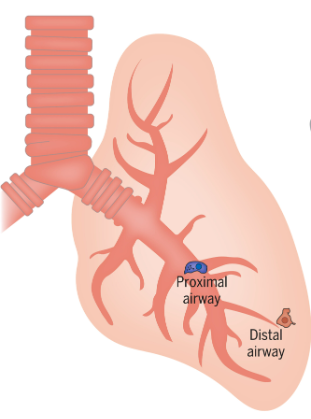
CONNECT
1.877.7CANCER
info@damonrunyon.org
One Exchange Plaza
55 Broadway, Suite 302
New York, NY 10006![]()
![]()
![]()
![]()
![]()